|
La sistemática filogenética, la filogeografía y la ecología
molecular son disciplinas encargadas de estudiar los procesos evolutivos de los organismos en diferentes niveles. En los últimos años han mostrado un avance notable propiciado por las innovaciones tecnológicas que han permitido disponer de una gran cantidad de información sobre la variabilidad de los seres vivos en forma de secuencias de adn y la aplicación de herramientas conocidas como marcadores moleculares. Aunado a esto, los avances teóricos han permitido emplear sofisticados algoritmos y herramientas estadísticas en el estudio de los procesos ecológicos y evolutivos. En particular, la aplicación de las herramientas moleculares ha revolucionado los marcos teóricos y metodológicos existentes en las ciencias biológicas, dando origen a marcos conceptuales que han permitido ubicar y responder mas recientes interrogantes. Los nuevos enfoques de investigación han sintetizado adecuadamente el surgimiento de innovadoras explicaciones a preguntas pendientes en evolución y ecología.
La amplitud y el tipo de problemas que se han respondido al combinar los enfoques de sistemática filogenética, filogeografía y ecología molecular son múltiples, con aplicaciones en aspectos taxonómicos, biogeográficos, ecológicos y de conservación biológica. La perspectiva integral en la comprensión de la historia evolutiva, los patrones de distribución geográfica y los procesos ecológicos de los seres vivos ha permeado las disciplinas evolutivas, provocando un fuerte impacto en términos del número de publicaciones, su relevancia e influencia en otras disciplinas biológicas.
El componente evolutivo
En el estudio de las relaciones filogenéticas y filogeográficas de los organismos van implícitos los procesos evolutivos, ya que involucran el estudio de las relaciones ancestrodescendiente y el reconocimiento de las causas históricas en la distribución geográfica de las especies. La ecología molecular también tiene un fuerte componente evolutivo al estudiar los cambios en la estructura genética de los organismos y los procesos asociados a tales cambios.
Los fundamentos de la sistemática filogenética se encuentran en las ideas de Charles Darwin, quien consideró que la evolución es producto de la descendencia con modificación a partir de un origen único de la vida, por lo que la historia evolutiva de los organismos debe reflejar sus patrones de ancestría y descendencia. El entomólogo Willi Hennig retomó las ideas de Darwin y fundó la sistemática filogenética o escuela cladística, una disciplina con más de cuarenta años de existencia que se ha enriquecido con propuestas teóricas y metodológicas a lo largo del tiempo. Su objetivo es inferir la historia evolutiva de los organismos con base en evidencias de los mismos (sus caracteres).
El principio de clasificación en cladística se basa en el concepto de homología, que hace referencia a dos estructuras morfológicamente semejantes y cuya semejanza se debe a que derivan de una estructura ancestral común. En cladística, las novedades evolutivas o los caracteres derivados compartidos, denominados sinapomorfías, son clave para inferir las relaciones evolutivas de los organismos. Las sinapomorfías permiten reconocer los grupos monofiléticos (entidades históricas con un origen común a todos sus descendientes). Los caracteres primitivos son denominados simplesiomorfías y son muy útiles para contrastar hipótesis de caracteres novedosos (sinapomorfías) contra las de caracteres primitivos (simplesiomorfías).
En cuestiones geográficas, Darwin tuvo dos grandes contribuciones: reconoció la intervención de causas históricas en la distribución de las especies y consideró que mediante las relaciones de ancestríadescendencia se pueden inferir las relaciones de las áreas geográficas. La filogeografía se enmarca dentro de la biogeografía histórica (el estudio de la dimensión espacial de la evolución biológica), cuya premisa es que “la Tierra y la biota evolucionan juntas”. Desde este enfoque evolutivo, una propuesta biogeográfica histórica representa un conjunto de hipótesis que se refieren a la distribución de la biota y sus interpretaciones históricas.
La filogeografía es un campo de investigación que surgió a finales de los ochentas, impulsada inicialmente por los trabajos del genetista evolutivo John C. Avise, quien acuñó la palabra Phylogeography y la definió como “el campo de estudio de los principios y procesos que gobiernan la distribución geográfica de los linajes genealógicos a nivel de especie o de especies cercanamente emparentadas”. Los análisis filogeográficos evidencian el grado de estructura poblacional de las especies. En aquellas especies con gran capacidad de movimiento (cetáceos, peces, iguanas marinas y aves rapaces), las estructuras filogeográficas suelen ser poco definidas por no ser muy divergentes. Especies con mucha menor movilidad (tortugas terrestres, serpientes, salamandras, roedores) suelen tener una estructura mejor definida; aunque es importante considerar que gran parte de la estructura filogeográfica de las especies puede ser explicada por el tamaño pequeño de las poblaciones y su limitado flujo génico, en lugar de una historia vicariante (proceso que divide la distribución geográfica de la biota debido a la formación de una barrera física que impide el flujo génico de los individuos).
El origen de la ecología molecular se encuentra en el trabajo de Edmund B. Ford de 1964, que marca el inicio de la aplicación de técnicas genéticas para responder a preguntas ecológicas. Dicho autor pone especial énfasis en “los ajustes y adaptaciones de las poblaciones a su ambiente desde una perspectiva que permita investigar el proceso real de la evolución que tiene lugar en el momento presente”. Éste fue el primer intento, con un enfoque evolutivo, de estudiar e interpretar las variaciones de la adecuación de los caracteres ecológicamente significativos de los organismos. A partir del desarrollo y uso de los marcadores moleculares (los puntos de referencia dentro del genoma) fue posible describir las variantes genéticas de los organismos y, por lo tanto, conocer las diferencias y similitudes genéticas entre individuos.
La ecología molecular es una disciplina que emplea herramientas conocidas como marcadores moleculares para resolver problemas ecológicos y evolutivos, abarcando el estudio de las relaciones genéticas a nivel de individuos, poblaciones y especies. Tiene un gran auge a partir de la publicación de la revista Molecular Ecology en 1992, cuando el término se hizo más popular, mostrando desde entonces un crecimiento constante en el número de publicaciones bajo este enfoque, con mayor diversidad de temáticas y conexión con otras disciplinas.
El ADN como fuente de información
El primer paso para realizar un estudio filogenético es determinar el tipo de caracteres que se emplearán, es decir, datos morfológicos o moleculares. Teóricamente un carácter molecular es lo mismo que un carácter morfológico (son atributos heredables de los organismos). Sin embargo, los caracteres moleculares presentan algunas ventajas con respecto de los caracteres morfológicos, como: 1) el enorme tamaño del conjunto de datos; 2) proporcionan un registro filogenético que abarca desde tiempos cercanos al origen de la vida hasta tiempos recientes; 3) en algunos casos no hay efecto ambiental en los caracteres (adn mitocondrial); 4) la descripción de los estados de caracteres no es subjetiva (esto es adenina o guanina, no “azul” o “menos azul”); 5) es relativamente fácil generar una matriz de miles de caracteres; 6) algunos genes homólogos existen en todos los grupos biológicos; 7) las diferentes regiones del genoma tienen distintas tasas de evolución; y 8) permiten estudiar problemas filogenéticos a distintos niveles.
Por su parte, los caracteres morfológicos presentan las siguientes ventajas: a) se pueden obtener a partir de ejemplares depositados en colecciones y museos; y b) se puede realizar el estudio de especímenes fósiles.
Para ambos tipos de carácter el tamaño de muestra es variable. Con los morfológicos, los taxónomos deben analizar muestras grandes para delimitar la variación de un carácter dentro de la especie, mientras que las muestras de los moleculares son usualmente más pequeñas. En general, no se puede argumentar cuál de los tipos de datos es mejor que el otro, ya que el tema se cierne a cómo se eligen y se registra la variación de los caracteres. Ante esto, la mejor opción es incorporar en los análisis ambos tipos de datos, lo cual resulta en mejores inferencias evolutivas que en aquellos que sólo incluyen un tipo de carácter.
Para determinar las relaciones genealógicas en los estudios filogeográficos es indispensable el uso de marcadores genéticos o secuencias de adn que no sean recombinantes. Las propiedades del adn de las mitocondrias que hacen que sea un marcador muy útil en filogeografía son las siguientes: 1) se hereda vía materna; 2) presenta una alta tasa de mutación, lo que implica sustituciones nucleotídicas; 3) no presenta recombinación genética; 4) al ser neutral, la distribución de los haplotipos (conjunto de alelos que provienen de un solo cromosoma) está más influenciada por los eventos demográficos en la historia poblacional que por la selección natural; y 5) se amplifica de manera relativamente sencilla.
En ecología molecular se emplean marcadores genéticos moleculares basados en adn (proteínas, secuencias) o en arn. Elegir el tipo de marcador molecular más adecuado depende de la pregunta que se quiere responder en el estudio ecológico. En términos generales, se debe considerar el nivel de variabilidad genética que puede revelar cada marcador. Asimismo se deben tomar en cuenta las características genéticas y evolutivas de los marcadores moleculares tales como: la forma parental de herencia (biparental o materna), la forma de herencia y expresión (dominante o codominante) y las tasas de mutación y de divergencia.
Los métodos de análisis
En sistemática filogenética, una vez que se ha determinado el tipo de caracteres (moleculares o morfológicos) se procede a decidir la forma en que se analizarán los mismos. En general, es posible incluir en el estudio el conjunto de caracteres por separado (congruencia taxonómica) o combinarlos en una matriz de caracteres única (evidencia total), la cual es una tendencia actual en los estudios publicados.
Para los análisis basados en congruencia taxonómica se utilizan árboles de consenso que se generan a partir de agrupamientos comunes entre las topologías mejor sustentadas y obtenidas a partir de diferentes matrices de datos. El procedimiento consiste en: 1) generar las matrices de datos; 2) obtener hipótesis de relación para cada conjunto de datos (conocidas como árboles fundamentales); y 3) realizar un consenso de las topologías obtenidas. Por su parte, los análisis basados en evidencia total utilizan la congruencia de caracteres para encontrar la mejor hipótesis filogenética a partir de un conjunto de sinapomorfías sin separarlas en tipos. Si se encuentran proposiciones igualmente parsimoniosas se usan consensos para resumir dichas alternativas, es decir, se busca una hipótesis única que explique de mejor forma los datos, lo que implica maximizar la congruencia de caracteres.
Cuando se ha decidido la forma en la que se analizarán los datos se procede a especificar el algoritmo bajo el cual se inferirán las relaciones filogenéticas. Los métodos más comúnmente utilizados en cladística son: máxima parsimonia, máxima verosimilitud y el análisis bayesiano.
El método de máxima parsimonia se basa en un mínimo de suposiciones a priori de los datos. Se asume que cualquier carácter heredable es una homología potencial. Por lo tanto, al momento de inferir los árboles filogenéticos todos los caracteres son tratados de igual manera, con el “mismo peso” o influencia. Bajo este criterio, el mejor árbol filogenético será aquel con la mínima cantidad de cambios evolutivos que se requieran para explicar una determinada matriz de caracteres.
Los métodos de máxima verosimilitud (maximum likelihood) y el análisis bayesiano son algoritmos estadísticos que se basan en modelos de evolución molecular y que toman en cuenta el conocimiento a priori de los caracteres, particularmente los moleculares (a saber, secuencias de nucleótidos de adn). La máxima verosimilitud estima cuál es la probabilidad de que la matriz de caracteres sea explicada por los árboles filogenéticos, mientras que el análisis bayesiano infiere cuál es la probabilidad de que los árboles filogenéticos sean bien explicados por los datos, es decir, la matriz de caracteres. Con el algoritmo de máxima verosimilitud se necesita calcular cada árbol posible que puede ser derivado de los datos según el modelo de evolución seleccionado. Además, se debe calcular la longitud de ramas para cada árbol diferente. Algunos autores prefieren utilizar el análisis bayesiano sobre la máxima verosimilitud porque utiliza “atajos” para los cálculos mediante un algoritmo conocido como cadenas Markov de Monte Carlo, con el cual se realizan búsquedas por medio de un número menor de árboles según sus valores de probabilidades posteriores. Esto permite que el análisis bayesiano demande menos poder computacional y sea más rápido que el de máxima verosimilitud.
En cladística, el cladograma (figura 1) resultante se interpreta como una hipótesis filogenética a partir de la cual, entre otras cosas, se pueden realizar propuestas de cambios taxonómicos o bien la interpretación de la evolución de los caracteres.
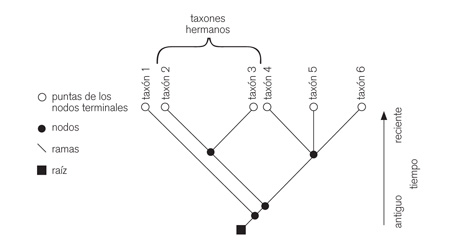 |
| Figura 1. Las relaciones genealógicas pueden ser representadas mediante árboles o cladogramas. Se representa la historia de los linajes y cómo estos han cambiado a través del tiempo. |
En filogeografía, los aspectos más relevantes a considerar en los análisis son: 1) los haplotipos de una sola especie o especies cercanamente emparentadas se pueden interpretar como una unidad terminal; 2) los datos que se analizan son secuencias de genes que generalmente provienen de alguna región del adn mitocondrial (a saber, región control, el citocromo A o B); 3) las relaciones genealógicas entre los haplotipos se representan por medio de filogramas, donde las longitudes de las ramas expresan la cantidad de cambios evolutivos o el grado de divergencia genética; 4) la construcción de los filogramas se basa en los mismos algoritmos de la sistemática filogenética; y 5) los filogramas se superponen a la distribución geográfica a fin de interpretar el proceso evolutivo responsable de la dispersión de la especie o las especies bajo estudio.
Para evaluar la robustez de las hipótesis de asociación entre estructura geográfica y la historia de las poblaciones se utilizan pruebas estadísticas. Entre las hipótesis filogeográficas se incluyen aquellas que analizan si la distribución geográfica de las especies se ha expandido posteriormente al Pleistoceno (hace 11 000 años) o si se ha contraído, permaneciendo actualmente aislada como refugios pleistocénicos. Están aquellas hipótesis que analizan la dirección prevaleciente del flujo génico que se da mediante la dispersión de los organismos en dos o más poblaciones separadas geográficamente y aquellas que examinan la contribución que han tenido los procesos históricos en la estructuración de la arquitectura genética de las poblaciones. También se han incorporado algunos fundamentos de la genética de poblaciones (deriva génica, procesos de migración, tamaño efectivo poblacional), lo que ha permitido diferenciar entre las distribuciones geográficas históricas y las expansiones de una distribución previa a una distribución menor. Como tal, se basa en un conjunto de criterios diferentes a los de la biogeografía histórica para aceptar o rechazar las hipótesis históricas.
En filogeografía también es posible inferir aspectos demográficos de las poblaciones cuando el tamaño de muestra lo permite. Estos análisis demográficos se basan en la teoría de la coalescencia, misma que proporciona los métodos para detectar eventos en el pasado de las poblaciones, tales como el incremento exponencial en tamaño. Con un tamaño poblacional grande, el tiempo de coalescencia entre dos secuencias de adn (tiempo en que ambas convergen en la secuencia que les dio origen) es más grande. En cambio, en poblaciones pequeñas el tiempo de coalescencia es menor, ya que pueden estar más cercanamente relacionadas. Con base en las distancias génicas entre los individuos y suponiendo cierta tasa evolutiva se pueden obtener estimados provisionales de tiempo absoluto de coancestría materna a partir de adn mitocondrial.
Los estimados de tiempo acumulados para muchos individuos dan como resultado frecuencias pareadas de distribuciones de tiempos de coancestría conocidas como distribuciones mismatch, que se presentan como histogramas en donde se muestra el número de diferencias de sitios nucleotídicos (o restricción) observados entre pares de haplotipos. El histograma es multimodal en poblaciones con equilibrio demográfico y unimodal en poblaciones con una reciente expansión demográfica. Los datos del histograma se pueden comparar con el modelo de una población en expansión demográfica. Con una prueba de 2 estadísticamente significativa se acepta que la diferencia entre ambas poblaciones no es debida al azar y se observa en la estructura de la gráfica si las poblaciones experimentaron recientemente un crecimiento exponencial.
Bajo el enfoque de ecología molecular se puede considerar el estudio de diversas preguntas o problemas ecológicos y evolutivos. Por lo tanto, además de los marcadores moleculares es necesario tener información de los cambios cuantitativos y cualitativos en la composición genética a nivel temporal (a lo largo de las generaciones) o espacial (entre individuos, poblaciones y especies). En ecología molecular se considera que las relaciones genéticas entre individuos, poblaciones y especies es la faceta que mejor define la naturaleza, por lo mismo esto establece la escala de los estudios bajo tal enfoque. En tal contexto, se pueden hacer comparaciones entre los padres y su progenie o entre individuos de la misma especie que se encuentran separados espacialmente.
Particularmente, por medio del estudio y la aplicación de los marcadores moleculares se pueden responder diversas preguntas que consideren la estructura y diversidad genética y las interacciones genéticas de los organismos con el medio ambiente; entre éstas se encuentran: ¿qué población dio origen a ciertos individuos?, ¿cuántas poblaciones existen?, ¿las poblaciones han expandido o contraído su distribución geográfica en el pasado reciente?, ¿difiere el tamaño de las poblaciones en el pasado con respecto del presente?, ¿cuáles son las relaciones genéticas de los individuos?, ¿cuáles individuos han migrado?, ¿cuáles individuos son clones?, ¿cuál es la distancia promedio de dispersión de los gametos?, ¿cómo influyen las características del paisaje en la estructura de las poblaciones?
Importancia de las disciplinas evolutivas
La importancia de los estudios de sistemática filogenética radica en los siguientes aspectos: a) los sistemas de clasificación filogenéticos son una forma de comunicación del conocimiento de la diversidad biológica; b) éstos proveen el fundamento teórico para el estudio comparativo de la diversidad y su conservación; y c) son el marco histórico para interpretar los patrones de similitudes entre los organismos, sus interacciones ecológicas y su distribución geográfica. En el mismo sentido, las filogenias constituyen la base teórica para estudios de especiación, ecología, biogeografía y más recientemente se han incorporado a los métodos de análisis de la filogeografía.
La filogeografía, por su parte, ha permitido avanzar en la descripción de las distribuciones geográficas de las especies, sus relaciones filogenéticas y las distancias genéticas entre linajes. Los estudios filogeográficos han logrado una mejor comprensión de la biogeografía regional y las áreas de endemismo. Al comparar los patrones filogeográficos de varios grupos biológicos en una misma región se puede comprender la historia regional, lo que se conoce como filogeografía comparada. Al mismo tiempo, entender las respuestas históricas de las especies a los cambios ambientales y la evolución de áreas evolutivamente aisladas ha sido muy importante para la conservación. Esto ha permitido establecer estrategias de manejo por debajo del nivel de especie con datos moleculares, una política de conservación conocida como Unidades evolutivamente significativas.
En el contexto de otras disciplinas, la filogeografía tiene un papel importante al funcionar como un enlace entre genética de poblaciones, sistemática filogenética y biogeografía. Este carácter de unir las disciplinas micro y macroevolutivas le confiere una posición muy destacada en la biogeografía histórica, permitiéndole abordar preguntas que tradicionalmente eran del interés de la ecología y la biología evolutiva al explorar los patrones de dispersión y la conectividad genética al interior de las poblaciones en ámbitos ecológicos actuales o muy recientes.
Finalmente, mediante el enfoque de la ecología molecular se amalgaman la teoría ecológica y la evolutiva. La utilidad de este enfoque radica en la forma en la que se pueden unir subdisciplinas ecológicas muy dispares desde una perspectiva evolutiva y abordar problemas que los actuales programas de investigación por sí solos simplemente no pueden.
Las soluciones a gran parte de las preguntas ecológicas actuales requieren un enfoque multidisciplinario que, en muchos casos, se puede basar en un marco molecular. Por lo tanto, es deseable que la mayoría de los ecólogos adquieran un conocimiento mínimo de los métodos moleculares con el fin de que puedan reconocerlos y elegir aquellos que podrían aplicar. La utilización de herramientas moleculares amplía la gama de problemas ecológicos que se pueden abordar, por lo que el futuro para la ecología molecular parece ser prometedor.
|





